

 We present the implementation of excited state Born–Oppenheimer molecular dynamics (BOMD) using a polarizable QM/MM approach based on a time-dependent density functional theory (TDDFT) formulation and the AMOEBA force field. The implementation relies on an interface between Tinker and Gaussian software and it uses an algorithm for the calculation of QM/MM energy and forces which scales linearly with the number of MM atoms.
We present the implementation of excited state Born–Oppenheimer molecular dynamics (BOMD) using a polarizable QM/MM approach based on a time-dependent density functional theory (TDDFT) formulation and the AMOEBA force field. The implementation relies on an interface between Tinker and Gaussian software and it uses an algorithm for the calculation of QM/MM energy and forces which scales linearly with the number of MM atoms.
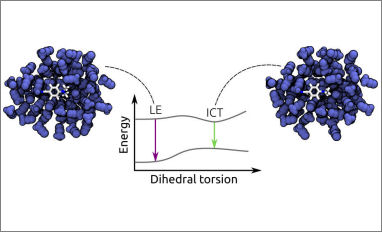
The resulting code can perform TDDFT/AMOEBA BOMD simulations on real-life systems with standard computational resources. As a test case, the method is applied to the study of the mechanism of locally-excited to charge-transfer conversion in dimethylaminobenzonitrile in a polar solvent. Our simulations confirm that such a conversion is governed by the twisting of the dimethylamino group which is accompanied by an important reorientation of solvent molecules.
The work is available at: https://doi.org/10.1039/D0CP03688A
